第24名方案(使用分子图和原子坐标)
在阅读了 DACON 的解决方案后,尤其是那个使用目标检测来检测原子和键坐标然后重建分子的方案,我一直在思考什么样的分子表示形式能更自然地被神经网络学习。
虽然直接预测 InChI 是最简单的解决方案,因为它需要最少的后处理,而且即使预测的字符串不是正确的 InChI,它也可能导致较低的 LD(Levenshtein 距离)。但它也有一些缺点:
- 它很长
- 它的原子编号非常复杂(并且对其非常敏感)
所以在训练之后,我很惊讶地发现神经网络实际上可以很好地学习直接预测正确的 InChI,且准确率很高。考虑到图像质量并不完美,LD < 2 对我来说似乎是非常好的结果。
虽然能够很好地预测 /c 层,但我发现它在识别立体化学(/t 层)方面很吃力,这本应是一个更简单的任务。如果它能预测整个原子连接序列,为什么不能只预测 + 或 - 呢?这不是最大的错误来源,因为如果其他层预测正确,它只会贡献 1-4 的 LD,但我仍然不清楚原因。
所以我开始寻找其他分子表示法来克服上述缺点。其中一种是 SMILES,它生成的字符串更短,另一种是分子图。我选择了后者。这并不完全是 DACON 第一名方案中所使用的(那是完全的目标检测方法,预测原子和键的坐标,然后根据原子和键的几何坐标确定哪些原子相连),相反,在我的表示中,我必须保留原子编号(尽管我将其从 InChI 编号改为更简单的基于原子 2D 坐标的编号——从左到右,从上到下),对于每个原子,除了其属性(如元素类型、手性、同位素、xy 坐标)外,我还预测了该原子与其他原子的连接——你可以将其视为邻接矩阵。
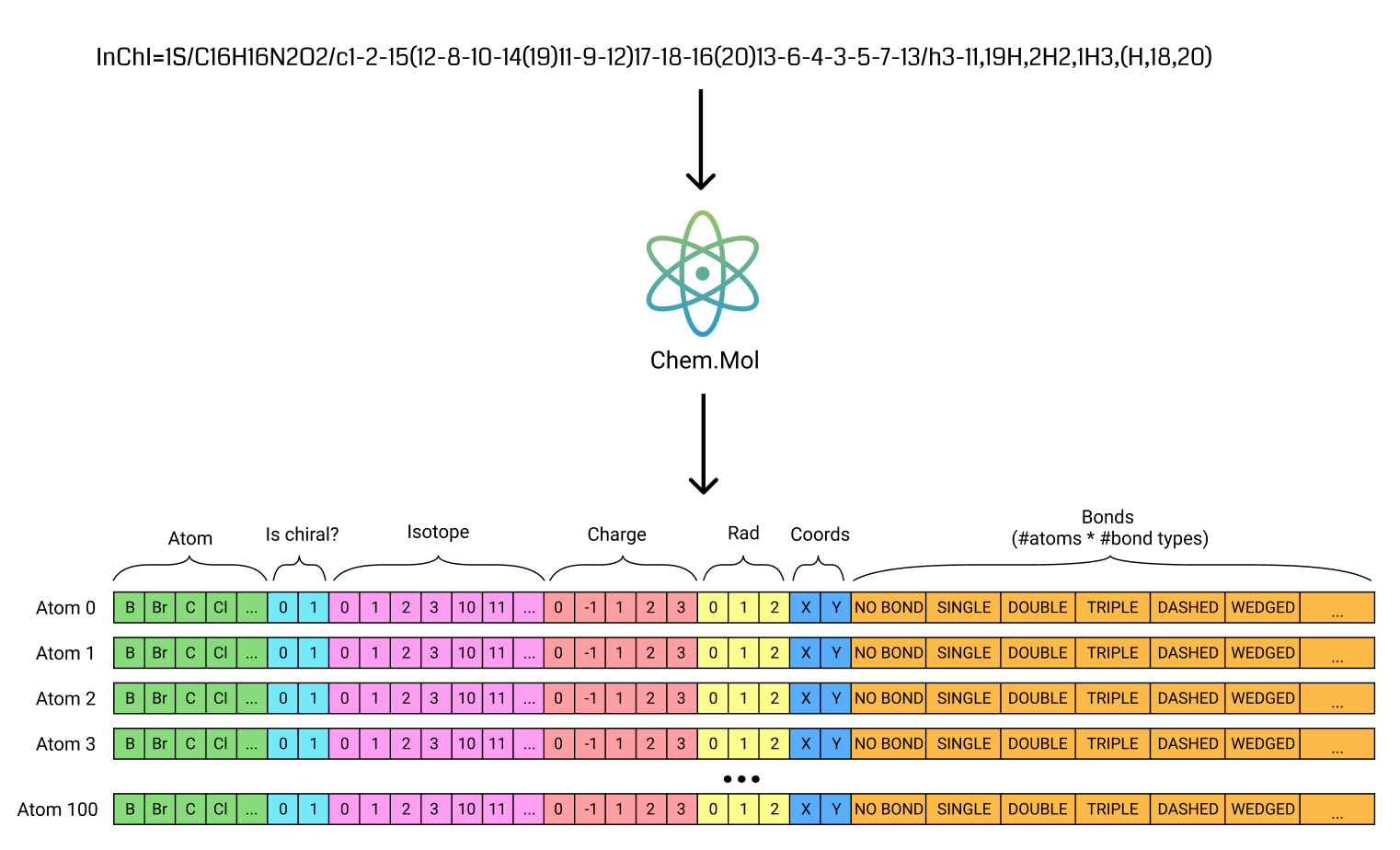
为了从原子属性和键恢复分子,我使用了类似下面的代码。这不是完整的代码,只是一个简单的例子,说明如何从头开始手动构建分子。这里正确检测立体化学的重要方法是 DetectBondStereochemistry 和 AssignChiralTypesFromBondDirs——它们实际上负责逆转渲染过程中发生的程序,即在计算分子坐标时,这些方法根据模型预测的原子 2D 坐标 分配 E/Z 和顺/反立体结构。
m = Chem.RWMol()
# 节点
m.AddAtom(Chem.Atom('C'))
m.AddAtom(Chem.Atom('C'))
m.AddAtom(Chem.Atom('C'))
m.AddAtom(Chem.Atom('C'))
m.AddAtom(Chem.Atom('F'))
m.AddAtom(Chem.Atom('O'))
m.AddAtom(Chem.Atom('Cl'))
# 边
m.AddBond(0, 1, Chem.BondType.SINGLE)
m.AddBond(1, 2, Chem.BondType.DOUBLE)
m.AddBond(2, 3, Chem.BondType.SINGLE)
m.AddBond(3, 4, Chem.BondType.SINGLE)
m.AddBond(3, 5, Chem.BondType.SINGLE)
m.AddBond(3, 6, Chem.BondType.SINGLE)
m.GetBondBetweenAtoms(3, 5).SetBondDir(Chem.BondDir.BEGINWEDGE)
# 坐标
coords = ((-2, -1), (-1, 0), (1, 0), (2, 1), (2, 0), (3.5, 1), (2, 2))
conf = Chem.Conformer(m.GetNumAtoms())
conf.Set3D(False)
for i, (x, y) in enumerate(coords):
conf.SetAtomPosition(i, (x, y, 0))
m.AddConformer(conf)
# 魔法步骤
Chem.SanitizeMol(m)
Chem.DetectBondStereochemistry(m)
Chem.AssignChiralTypesFromBondDirs(m)
Chem.AssignStereochemistry(m)
m.Debug()
opts = Draw.MolDrawOptions()
opts.addAtomIndices = True
opts.addStereoAnnotation = True
Draw.MolToImage(m, options=opts)
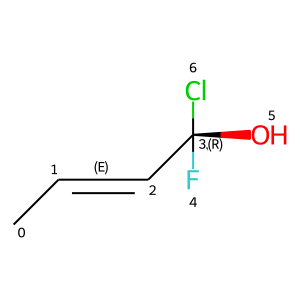
与此同时,我也在寻找用尽可能接近原始训练图像的其他图像来