斯坦福Ribonanza RNA折叠比赛第44名解决方案
上下文部分
- 业务背景:https://www.kaggle.com/competitions/stanford-ribonanza-rna-folding/overview
- 数据背景:https://www.kaggle.com/competitions/stanford-ribonanza-rna-folding/data
方法概述
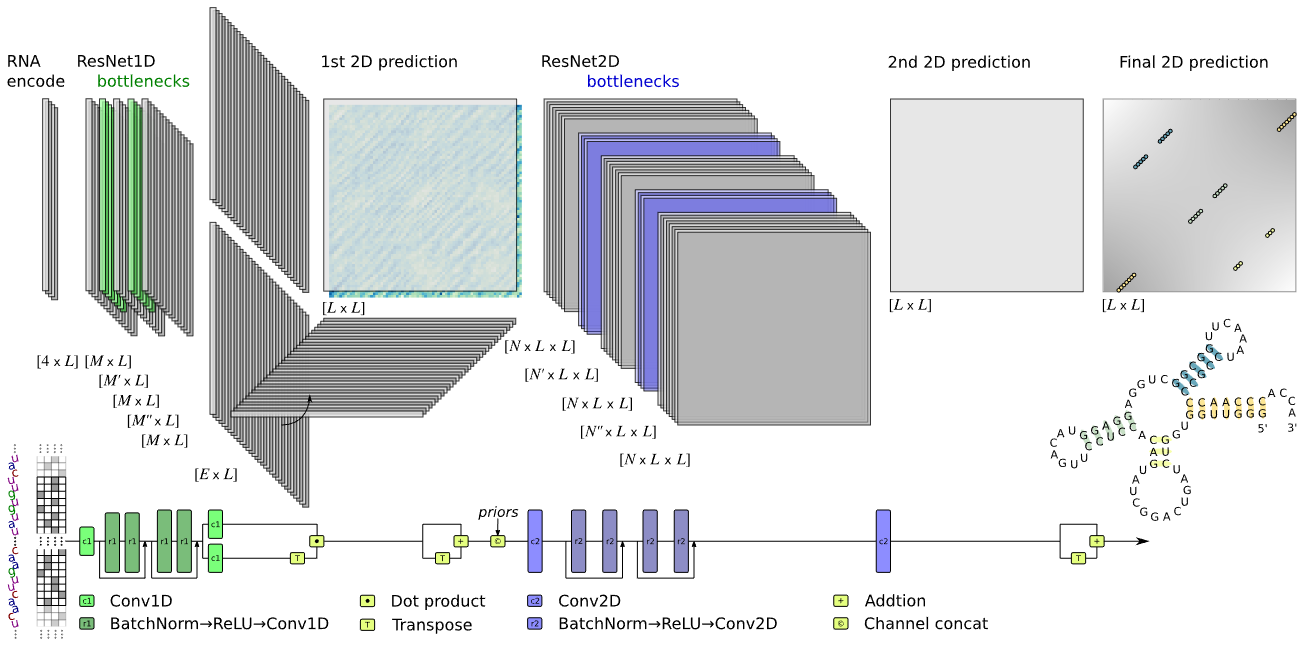
这是我的第一篇总结(也是第一次进入奖牌区 😁)。感谢组织者和参与者!我的方法基于我们实验室在RNA二级结构预测方面的工作[1](该文章包含源代码)。这是一个基于ResNet的模型,将输入序列作为独热编码表示,并输出相互作用矩阵预测(类似于提供的bpp)。该模型首先进行一维特征提取,然后通过简单的矩阵乘法转换为二维,并经历第二阶段的二维特征提取以达到最终预测。对于本次比赛,输出通过按列求和"展平",从而得到序列中每个核苷酸激活的表示。内部的二维表示允许我们添加bpp信息作为额外的通道来学习特征。
在数据方面,这是一个相对简单的方法,最佳模型使用了SNR>0.5的训练数据。训练/测试分割使用cdhit-est(80%阈值)[2]对序列进行聚类。这对于避免在相似序列上过拟合模式非常重要。同时使用了提供的BPP。
提交细节
我想尝试不同的方法,但重点改进上述详细描述的方法。由于训练耗时,使用每个序列聚类的中心点被证明可以达到有竞争力的结果(与我的最佳提交相比),因此这是开发过程中的方法。最好能过滤泄露的测试序列,但似乎我的公共排行榜上的最佳解决方案在私有排行榜上是一样的,所以对模型来说是好消息。最终结果平均了5个模型,其中3个只使用了中心点,因为我时间不够了。此外,除了查看公共排行榜外,没有使用测试数据做任何事情。
一个有趣的观点是使用BPP信息。由于它基于经典的RNA结构预测方法,不模拟假结,因此在假结情况下可能会影响模型预测。我训练了不使用BPP的模型,得到的平均结果要差得多(公共排行榜约0.16)。我还没有在私有排行榜上尝试,但如[3]所述分析一个样本案例,可以看到最终集成模型(中间图像)错过了参考预测(上方图像)中箭头指向的预测,而不使用BPP的模型(底部图像)则有一些相似之处。

没有效果的方法:
- 尝试了白噪声和翻转增强
- 使用SNR小于0.5的序列
- 尝试在训练中使用错误,但没有得到收敛的方法。不过这可能有效
希望看到更多关于生物序列的工作!
来源
[1] https://www.biorxiv.org/content/10.1101/2023.10.10.561771v1
[2] https://sites.google.com/view/cd-hit
[3] https://www.kaggle.com/competitions/stanford-ribonanza-rna-folding/discussion/444653